

Author: Ishika Jaitly
Mitochondria are cellular substructures that function as energy powerhouses for almost all eukaryotic cells, including those of humans [1]. The number of mitochondria in a cell can vary widely [2]. Red blood cells, for example, have none, whereas liver cells can contain up to two thousand [2]. Mitochondria are integral to our survival due to the variety of roles they play in our biochemistry, the most important of which is the production of adenosine triphosphate (ATP) by aerobic respiration [1]. This process involves oxidative phosphorylation, which utilizes oxygen to release the energy contained in the foods we eat. The energy is initially stored in ATP, which subsequently releases this energy to power the body’s metabolic reactions. During this process, ATP is broken down into adenosine diphosphate (ADP) and inorganic phosphate (Pi).
Since mitochondria are essential to our very existence, it is unsurprising that the effects of mitochondrial diseases can be severe, and sometimes lethal, spanning a wide-ranging spectrum of physiological and biochemical symptoms [1,3,4] (see Table 1 for examples). Mitochondrial diseases in humans can arise from a myriad of mutations in mitochondrial and nuclear (cellular) DNA [4]. Collectively, mitochondrial diseases are reported to affect 1 in every 5,000 adults, making them quite prevalent in comparison to other hereditary metabolic disorders [4].
| Primary Mitochondrial Disease Subtype | Classic Symptoms |
| Leigh Syndrome (subacute necrotizing encephalomyelopathy) | Muscle spasms, fatigue [5]. |
| Alpers–Huttenlocher syndrome | Epilepsy, liver deterioration [6]. |
| Leber hereditary optic neuropathy | Progressive loss of vision in both eyes, most commonly seen in young males [1]. |
| Mitochondrial encephalopathy lactic acidosis and stroke-like episodes (MELAS) | Seizures and intermittent occurrences of headache and vomiting [7]. |
Table 1. Some well-known clinically defined primary mitochondrial diseases and their respective symptoms.
Mitochondrial diseases are characterized by both a diminished capacity to produce ATP as well as an elevated production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) [3]. Commonly known as free radicals, ROS and RNS are naturally produced during oxidative phosphorylation and serve an essential role as signalling molecules in the normal functioning of a cell [4]. However, a dysregulated increase in the level of these free radicals, as seen in mitochondrial diseases, can result in oxidative damage by overwhelming the cellular antioxidant defence mechanism, of which glutathione is the principal component [4]. The consequent depletion of glutathione [3] impedes the efficient quenching of free radicals, leading to redox imbalance and oxidative stress [4]. This, in turn, can compromise the structural and functional integrity of the cell’s nucleic acids, proteins and lipids [4]. As a result, the activity of the electron transport chain (ETC) is hindered, decreasing ATP production during oxidative phosphorylation [8]. Since the cellular synthesis of glutathione is ATP-dependent, decreased ETC activity also undermines glutathione synthesis, further exacerbating oxidative stress in a vicious cycle [8]. Hence, mitochondrial diseases present as a two-front attack on cellular glutathione levels; the excess free radicals deplete glutathione reserves, and the decreased ATP production slows glutathione replenishment, ultimately resulting in chronic glutathione deficiency [8].
The appreciably high incidence of mitochondrial diseases highlights a growing need to devise effective therapeutic strategies. With the current absence of technologies to rectify the actual mutations that underlie mitochondrial diseases, medical science can only address the resulting oxidative stress as a potential therapeutic option. Therefore, therapy primarily focuses on providing patients with supportive care to manage their symptoms using dietary supplements [9]. Commonly prescribed or recommended dietary supplements include coenzyme Q10 and vitamin B, C, E and K [9,10]. Unfortunately, no concrete evidence of their efficacy exists to support their beneficial use in addressing the symptoms of mitochondrial diseases [9, 10].
Given the strong association between glutathione depletion and the progression of mitochondrial diseases, dietary supplements with the potential to increase intracellular glutathione reserves have been the subject of much research in recent years [11]. One of the prime candidates has been N-acetylcysteine (NAC), which is the gold standard antidote for acute acetaminophen (paracetamol) overdose. In this particular case, cysteine is severely depleted, and NAC helps to restore cellular glutathione levels to homeostasis by providing a source of cysteine. However, no clinical trials have been reported that provide any evidence of its effectiveness for raising the chronically low homeostatic level of glutathione that occurs in primary mitochondrial diseases [1, 11]. Additionally, in the context of lowered ATP production, which is a hallmark of mitochondrial diseases, it is important to note that NAC, essentially only being a form of cysteine, will require 2 ATP molecules to be converted to glutathione. This is because cysteine is at the very beginning of the two-step cellular synthesis of glutathione (see Figure 1). For a detailed explanation, read our article on NAC https://www.glutathionereporter.com/n-acetylcysteine-nac-cannot-increase-cellular-glutathione-levels/.
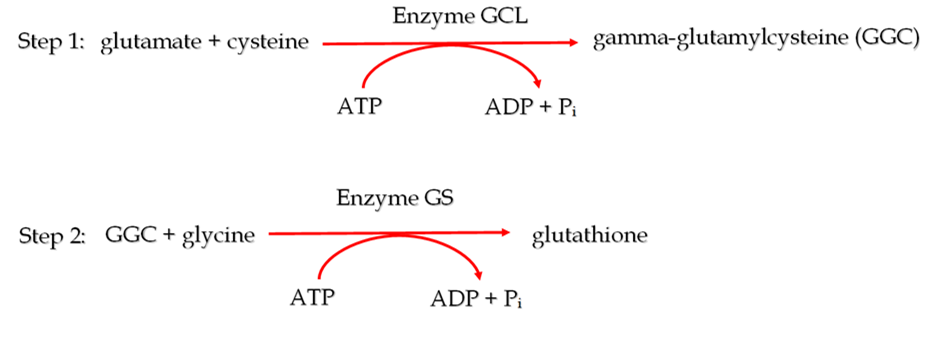
Figure 1. The two sequential ATP-dependent reactions by which glutathione is synthesized inside cells, with the enzyme catalyzing the reaction and the number of ATP molecules required for the reaction to occur, displayed above and below the arrows respectively.
Unlike NAC, dietary supplementation with gamma-glutamylcysteine (GGC), the immediate precursor to glutathione, can elevate intracellular glutathione levels above homeostasis [12]. This increased glutathione pool may help contend with the increased flow of free radicals from dysfunctional mitochondria. Moreover, the conversion of GGC to glutathione only requires 1 molecule of ATP as it is the product of the first step in glutathione synthesis, as shown in Figure 1. This low energy feature of GGC may be particularly useful for overcoming the ATP inadequacy that occurs in mitochondrial diseases. Finally, GGC may be capable of acting as an antioxidant in its own right by removing key ROS such as hydrogen peroxide and superoxide anions independently of glutathione [13]. This may be advantageous in severe cases of mitochondrial diseases where there could be insufficient ATP available to synthesize glutathione from GGC.
In summation, GGC’s potential to elevate intracellular glutathione levels above homeostasis in an energy-efficient manner, makes it a promising candidate as a potential therapeutic for mitochondrial diseases.
Reference
- Schapira, AHV 2006, ‘Mitochondrial Disease’, The Lancet, vol. 368, no. 9529, pp. 70-82.
- Jones, M, Fosbery, R, Gregory, J, Taylor, D 2014, Cambridge International AS and A Level Biology Coursebook, 4th edn, Cambridge University Press, United Kingdom.
- Atkuri, KR, Cowan, TM, Kwan, T, Ng, A, Herzenberg, LA, Herzenberg, LA, Enns, GM 2009, ‘Inherited disorders affecting mitochondrial function are associated with glutathione deficiency and hypocitrullinemia’, Proceedings of the National Academy of Sciences, vol. 106, no. 10, pp. 3941-3945.
- Enns, GM, Moore, T, Le, A, Atkuri, K, Shah, MK, Cusmano-Ozog, K, Niemi, AK, Cowan, TM 2014, ‘Degree of Glutathione Deficiency and Redox Imbalance Depend on Subtype of Mitochondrial Disease and Clinical Status’, The Public Library of Science One, vol. 9, no. 6.
- Baertling, F, Rodenburg, RJ, Schaper, J, Smeitink, JA, Koopman, WJH, Mayatepek, E, Morava, E, Distelmaier, F 2014, ‘A guide to diagnosis and treatment of Leigh syndrome’, Journal of Neurology, Neurosurgery & Psychiatry, vol. 85, no. 3, pp. 257-265.
- Saneto, RP, Cohen, BH, Copeland, WC, Naviaux, RK 2013, ‘Alpers-Huttenlocher Syndrome’, Pediatric Neurology, vol. 48, no. 3, pp. 167-178.
- El-Hattab, AW, Adesina, AM, Jones, J, Scaglia, F 2015, ‘MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options’, Molecular genetics and metabolism, vol. 106, no. 1-2, pp. 4-12.
- Hargreaves, IP, Sheena, Y, Land, JM, Heales, SJR 2005, ‘Glutathione deficiency in patients with mitochondrial disease: Implications for pathogenesis and treatment’, Journal of Inherited Metabolic Disease, vol. 28, no. 1, pp. 81-88.
- Chinnery, PF, Turnbull, DM 2001, ‘Epidemiology and treatment of mitochondrial disorders’, American Journal of Medical Genetics, vol. 106, no. 1, pp. 94-101.
- Gorman, GS, Chinnery, PF, DiMauro, S, Hirano, M, Koga, Y, McFarland, R, Suomalainen, A, Thorburn, DR, Zeviani, M, Turnbull, DM 2016, ‘Mitochondrial diseases’, Nature Reviews Disease Primers, vol. 2, no. 26080, pp. 1-22.
- Enns, GM, Cowan, TM 2017, ‘Glutathione as a Redox Biomarker in Mitochondrial Disease – Implications for Therapy’, Journal of Clinical Medicine, vol. 6, no. 5, pp. 50.
- Zarka, MH, Bridge, WJ 2017, ‘Oral administration of γ-glutamylcysteine increases intracellular glutathione levels above homeostasis in a randomised human trial pilot study’, Redox Biology, vol. 11, pp. 631-636.
- Quintana-Cabrera, R, Fernandez-Fernandez, S, Bobo-Jimenez, V, Escobar, J, Sastre, J, Almeida, A, Bolaños, JP 2012, ‘γ-Glutamylcysteine detoxifies reactive oxygen species by acting as glutathione peroxidase-1 cofactor’, Nature Communications, vol. 3, no. 718, pp. 1-8.
Very well written and easy to understand the concepts. Keep it up.
An interesting read, very well written & explained. Keep up the good work. Am sure it will be well received.